Condroblastoma
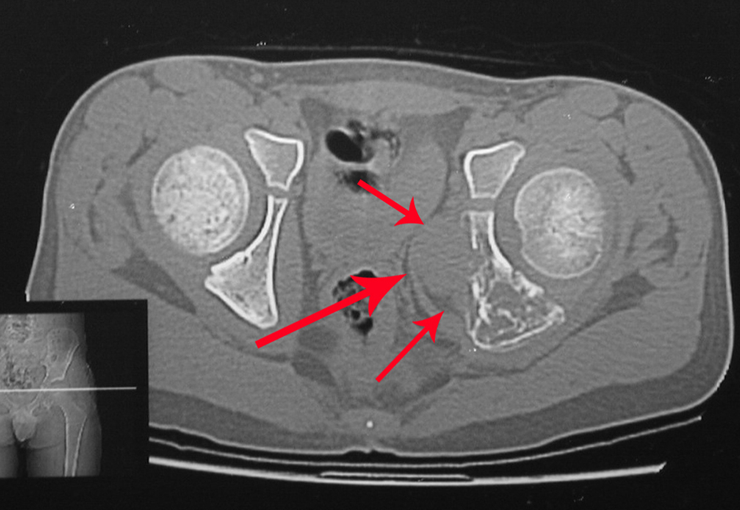
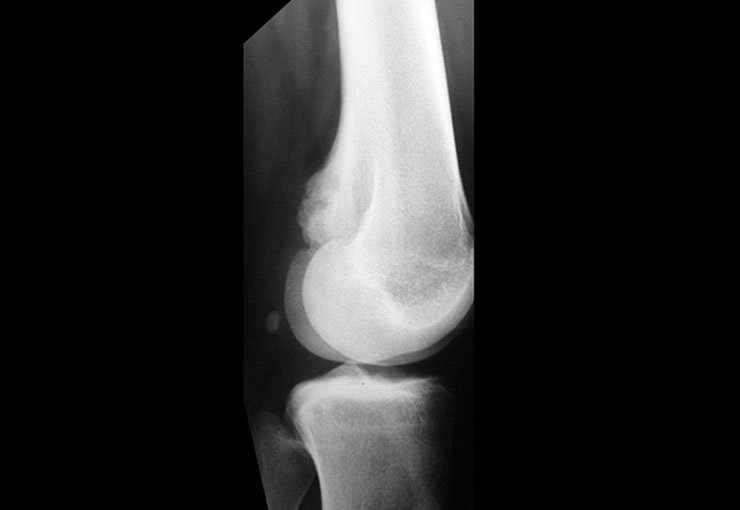
O condroblastoma é uma neoplasia benigna rara que corresponde a aproximadamente 1,8% de todos os tumores ósseos. Este tipo de tumor tem preferência pela epífise dos ossos longos e geralmente se manifesta como uma lesão de rarefação óssea, com focos de calcificação. É mais comum em pacientes do sexo masculino, ocorrendo tipicamente durante a primeira e segunda décadas de vida, quando a placa de crescimento ainda está aberta.
Descrito inicialmente por Codman em 1931, o condroblastoma foi inicialmente associado ao “tumor de células gigantes calcificado” do úmero proximal. No entanto, estudos posteriores mostraram que se tratava de uma entidade tumoral distinta do tumor gigantocelular (TGC).
Devido à sua localização intra-articular, o condroblastoma pode apresentar sintomas semelhantes aos da artrite. Além disso, pode demonstrar agressividade local, causando erosão da cortical óssea, da placa de crescimento e invasão articular. Frequentemente, áreas de cistos ósseos aneurismáticos podem estar associadas a manifestações radiográficas de agressividade local.
O tratamento do condroblastoma geralmente envolve a curetagem intralesional seguida de adjuvantes locais, como fenol, eletrotermia ou nitrogênio líquido, além da colocação de enxerto ósseo autólogo ou cimento de polimetilmetacrilato. Em casos mais avançados, pode ser necessária a ressecção segmentar seguida de reconstrução com prótese ou artrodese em casos recidivados ou muito avançados.
O prognóstico do condroblastoma pode ser reservado devido ao risco de recidiva local e às possíveis complicações ortopédicas, incluindo degeneração articular e déficit de crescimento.
Embora raramente, o condroblastoma pode levar a metástases pulmonares com histologia semelhante à do tumor benigno, sem apresentar atipias. O tratamento para essas metástases pode ser o acompanhamento clínico e de imagens e se a observação indicar evolução pode se fazer necessária a exérese cirúrgica.
Em resumo, o condroblastoma é uma neoplasia óssea benigna que, embora rara, requer atenção cuidadosa devido à seu potencial de agressividade local e possíveis complicações a longo prazo. O diagnóstico precoce e o tratamento adequado são essenciais para garantir o melhor prognóstico possível para os pacientes afetados.
veja o caso de Condroblastoma do Fêmur.